|
If your email program has trouble displaying this email or images, view as a webpage.
|
|
|
Back to TOP
 FDA Signs Mutual Recognition Agreement with Swissmedic
Swissmedic, the Swiss authority responsible for the authorization and supervision of therapeutic products, is the latest regulatory authority to sign an agreement with the FDA allowing the two regulators to share documents from routine good manufacturing practice (GMP) inspections of pharmaceutical manufacturing facilities.
The Agreement on Mutual Recognition between the Swiss Confederation and the United States of America Relating to Pharmaceutical Good Manufacturing Practice, signed by the FDA and Swissmedic in Washington on January 12, is intended to reduce unnecessary costs and duplicative efforts
The FDA already has a Mutual Recognition Agreement (MRA) in place with the European Union and another one with the United Kingdom. The MRA with the EU covers GMP inspections of facilities making human drugs. The MRAs with the UK and Switzerland cover GMP inspections of both human and animal drug manufacturing facilities.
The 15-page MRA with Switzerland includes provisions with regard to:
- What inspections and products are covered by the MRA.
- When one regulator intends to accept official GMP documents from the other.
- How the regulators will transmit official GMP documents to one another.
- Under what circumstances each regulator expects to request that the other conduct inspections.
- Under what circumstances a regulator may suspend the other for purposes of the MRA.
- The establishment of two different committees to facilitate the effective functioning of the MRA.
The FDA’s Office of Global Policy and Strategy and the Office of the U.S. Trade Representative (USTR) have been negotiating the U.S.-Switzerland MRA for several years. Before the MRA enters into force, the FDA must determine whether Swissmedic is capable of conducting inspections that meet U.S. requirements, and Swissmedic must make a similar determination with respect to the FDA meeting Swiss requirements.
 Seated from left to right are the signatories: FDA Deputy Commissioner for Policy, Legislation, and International Affairs Andi Lipstein Fristedt; Deputy U.S. Trade Representative Jayme White; State Secretary for the State Secretariat for Economic Affairs (SECO) Helene Budliger Artieda; Swissmedic Head of Management Services and International Affairs Dr. Jörg Schläpfer. Standing in the back from left to right: Sloane Strickler (USTR), Jade Pham (FDA), Sarah Short (USTR), Jason Buntin (USTR), and Christoph Schelling (SECO).
“In today’s global pharmaceutical market, MRAs offer a way for the FDA to work more efficiently and maximize its resources,” said Andi Lipstein Fristedt, FDA Deputy Commissioner for Policy, Legislation, and International Affairs, who signed the agreement on behalf of the FDA. “Once the MRA enters into force, the FDA will be able to rely on the factual findings of Swissmedic experts in many cases, thus avoiding duplicate inspections and allowing the FDA to expand its inspectional reach,” she said.
Having MRAs in place with the EU and the UK was especially helpful during the height of the COVID-19 pandemic when COVID-related travel restrictions limited the FDA’s ability to inspect sites. Instead of traveling, the FDA could receive and rely on inspection reports from the EU or the UK, including inspections conducted both within and outside their countries.
For example, during FY2021, the FDA reviewed and classified 139 site inspections in 18 MRA partner countries and six other countries, according to a report from the FDA’s Office of Pharmaceutical Quality.
Adding an MRA with Swissmedic is an important next step for the FDA since Switzerland is considered a global hub for pharmaceutical manufacturing. Two major pharma multinational corporations, Roche and Novartis, are based in Switzerland and the country also boasts a variety of small- and medium-sized multinational companies.
“This MRA will help streamline the movement of pharmaceutical goods and is a step in the right direction to create a safer, stronger, more reliable supply chain that minimizes drug shortages,” said Deputy U.S. Trade Representative Jayme White, who signed the document on behalf of USTR.
Swiss State Secretary for the State Secretariat for Economic Affairs Helene Budliger Artieda, and Swissmedic Head of Management Services and International Affairs Dr. Jörg Schläpfer signed the document on behalf of Switzerland.
FDA Serves Up Food Traceability Final Rule
To help facilitate faster identification and rapid removal of potentially contaminated food from the market, in November the FDA issued the final rule on Requirements for Additional Traceability Records for Certain Foods, or the Food Traceability Final Rule for short, to help reduce the occurrence of foodborne illnesses and deaths.
The final rule aligns with current industry best practices and covers the entire food supply chain including retail food establishments, restaurants, farms, and both domestic and foreign entities handling food for U.S. consumption. With this final rule, the FDA is taking a huge step forward for traceability efforts by identifying and standardizing the information necessary for the effective and rapid tracing of food.
The final rule will apply to those foods included on the FDA’s Food Traceability List (FTL). This list includes, among other things, certain fresh fruits and vegetables, a variety of soft/semi-soft cheeses, deli salads, and some categories of seafood, as well as foods that use these foods as an ingredient in the same form (e.g., fresh). The FTL was developed through a Risk Ranking Model, a process that included Federal Register solicitation of public input, refinement based on that input, and peer review.
Additional record-keeping across the food supply chain is at the core of the final rule. Companies that manufacture, process, pack, or hold FTL food must keep records relating to specific activities (Critical Tracking Events, or CTEs) they perform with respect to those foods.
These include:
- Harvesting.
- Cooling (before initial packing).
- Initial packing of a raw agricultural commodity other than a food obtained from a fishing vessel.
- First land-based receiving of a food obtained from a fishing vessel.
- Shipping.
- Receiving.
- Transformation of the food. This involves manufacturing/processing or changing a food (e.g., by commingling, repacking, or relabeling) or its packaging or packing, when the output is a food on the FTL.
For each CTE, the covered entity must keep Key Data Elements (KDEs), which will vary depending on the CTE being performed.
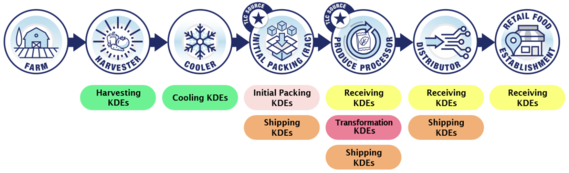 Example CTEs and associated KDEs for fresh cucumbers.
Central to the proposed requirements is the assignment, recording, and sharing of traceability lot codes (TLCs) for FTL foods, as well as linking these TLCs to other information identifying the foods as they move through the supply chain.
Prior to the final rule, KDEs were not standardized, and those involved in the supply chain were not required to share specific information (such as the newly-mandated TLC and where the TLC was assigned — the “TLC Source”). This created a system in which it has been necessary to take one step forward to identify where the food has gone and one step back to identify the previous source, and has made it difficult to rapidly track and trace food in the event of an outbreak. This can slow down the investigation, potentially resulting in additional sickness or death, overly broad recalls, millions of dollars in avoidable product loss, and a loss of consumer trust.
The final rule specifies that in the event of an outbreak or other threat to the public health, covered entities must provide the required information to the FDA in an electronic sortable spreadsheet within 24 hours of the FDA's request, or within some reasonable time to which the FDA agrees.
The FDA recognizes that entities, including the many foreign entities who export food to the United States, will have to work closely with their supply chain partners as they come into compliance with the final rule. The agency has set January 20, 2026, as the compliance date for the record-keeping requirements. This time frame will allow affected companies to learn about the final rule and to put in place their plan and processes for how they track and share information with other entities in their supply chain.
As part of the agency’s activities related to the final rule, the FDA intends to conduct specific outreach and education activities for entities in foreign countries to help them understand their responsibilities under the new rule. The FDA’s foreign offices in China, Europe, India, and Latin America — part of the agency’s Office of Global Policy and Strategy — will be important players in providing such outreach. These activities will include resource materials in key languages; webinars and other training efforts in collaboration with regional partners; and targeted outreach and engagement with specific regional government and industry stakeholders. The final rule also provides full and partial exemptions for covered entities in specific circumstances that may apply to foreign firms.
 As part of the final rule, affected entities will need to establish and maintain a Traceability Plan that covers the following information:
- The procedures used to maintain the records required under this rule, including the format and location of these records.
- The procedures used to identify foods on the FTL that the entity manufactures, processes, packs, or holds.
- How traceability lot codes are assigned to foods on the FTL, if applicable for that company.
- Identification of a point of contact for questions regarding the entity’s traceability plan and records.
- For those entities that grow or raise a food on the FTL (other than eggs), a farm map showing the areas in which those foods are grown or raised.
The rule also requires that records be kept as original paper or electronic records or true copies. All records must be legible and stored to prevent deterioration or loss.
The Food Traceability Final Rule is a key component of the FDA’s New Era of Smarter Food Safety Blueprint and implements Section 204(d) of the FDA Food Safety Modernization Act (FSMA). Tech-enabled traceability is one of the four core elements of the New Era Blueprint and is of key importance to the agency’s approach to food safety now and into the coming decades. As has been seen with outbreaks in fresh leafy greens and other foods over the past decade, the anonymity of one’s extended supply chain and lack of traceability in the food system are vulnerabilities that hinder more significant progress in rapid traceback efforts to identify contaminated foods. It also stands in the way of the transparency needed to better understand the supply chain in the event of public health crises, such as the COVID-19 pandemic, and to create a more nimble, resilient, and interoperable food system.
Accompanying the drive toward greater transparency through strong record-keeping is the now widely shared understanding that the foundation for reducing the risk of preventable foodborne illness in today’s global food system is action by the food industry. Food managers across the farm-to-table spectrum are called upon to manage their operations and supply chains in a manner that provides documented assurances that appropriate identification and tracking measures are being implemented as a matter of routine practice every day. Such robust traceability will transform food safety across the supply chain.
Outlook is Bright for Program That Encourages a Global Approach to Medical Device Auditing
One of the most visible, if not most successful programs to emerge from a decades-old drive to harmonize medical device regulation across the globe is the 5-year-old Medical Device Single Audit Program (MDSAP).
The program allows participating companies to satisfy the quality system requirements of multiple regulatory jurisdictions with a single audit. Audits are conducted at least annually by third-party auditing organizations, which have been recognized by participating regulatory authorities. MDSAP is the brainchild of the International Medical Device Regulators Forum (IMDRF), a voluntary group of medical device regulators who seek to accelerate international medical device regulatory harmonization and convergence, building on the foundations of the earlier Global Harmonization Task Force.
At its inaugural meeting in Singapore in 2012, the IMDRF identified a work group to develop specific documents for advancing MDSAP. After a successful pilot, MDSAP was established as a permanent program in 2017. About half of the IMDRF membership participates in MDSAP, including regulatory authorities in five countries: the Therapeutic Goods Administration of Australia; Brazil’s Agência Nacional de Vigilância Sanitária; Health Canada; Japan’s Ministry of Health, Labour and Welfare; the Japanese Pharmaceuticals and Medical Devices Agency; and the FDA. Europe does not participate in the program.
Since the program’s founding, it has enjoyed continued growth, according to data cited last month by Associate Commissioner for Global Policy and Strategy Mark Abdoo at the MDSAP Forum, an annual meeting of MDSAP participants.
 In MDSAP’s first year, there were only 229 registered medical device manufacturing sites, Abdoo said. However, he noted that by the third quarter of 2022, that number had grown to 6,459 sites located throughout the world. While a high percentage of these sites (42%) are located in the United States, a significant portion of sites are located elsewhere, including Germany (8%), Canada (7%), China (6%), and Japan (4%). In addition, the United Kingdom, France, Switzerland, and South Korea each account for 3% of the MDSAP sites, while Italy and Israel account for 2% of them. However, the data doesn’t indicate how many device companies participate.
Over the years, third-party auditing organizations have conducted a total of 16,612 audits under the program with nearly one-half taking place during the COVID-19 pandemic. From March 11, 2020, to November 17, 2022, a total of 8,882 audits were submitted. Sixty-two percent of those contained a remote component.
Abdoo said it’s not surprising that the program has seen substantial growth “given its many benefits.” They include a reduced regulatory burden for industry, more efficient and flexible use of regulatory resources, greater transparency, and “last, but not least, they benefit patient health and patient access as a result of ease of entry to multiple markets.”
Canada has certainly contributed to the escalating interest in MDSAP, he added. In 2019, the country began requiring audits via MDSAP for all manufacturing facilities with Class II, III, and IV medical device licenses.
MDSAP’s original intent — promoting globally a greater alignment of regulatory approaches and technical requirements based on international standards and best practices — has also affected FDA policy. MDSAP’s auditing organizations use the international consensus standard ISO 13485, a standard used by many regulatory authorities around the world but not used by the FDA. That is now changing. In 2021, the FDA announced a proposed rule to its medical device quality requirements that would more closely align to the standard, once the rule is finalized.
“These are positive times for MDSAP,” Abdoo said. “Given MDSAP’s many benefits and the support of all of you, we can foresee continued adoption of MDSAP globally, which will promote consistency in the regulation of devices and provide timelier introduction of safe, effective, high-quality devices for patients.”
|
|
|
Back to TOP
 FDA Launching Joint Premarket Pilot Program with Health Canada
On January 11, the FDA announced it was partnering with Health Canada to launch a pilot program to test whether it is feasible for medical device sponsors to use a specialized online template for jointly filing a premarket submission with both agencies.
The template is formally known as the “electronic Submission Template and Resource,” or eSTAR for short. It is an interactive PDF form that guides applicants through the process of preparing a comprehensive medical device submission, and was first introduced by the FDA’s Center for Devices and Radiological Health (CDRH) in February 2020 as an alternate method for industry to prepare 510(k) notifications.
 In 2022, eSTAR was updated to include content for both Health Canada’s Class III and Class IV applications as well as the FDA’s De Novo and PMA submissions.
eSTAR is complementary to CDRH internal review templates, thereby allowing the agency to receive information and evaluate the submission more efficiently and consistently.
CDRH has been working to implement policies and programs that promote global regulatory alignment, and the pilot is very much in keeping with that effort. The eSTAR template follows the recommended structure of submission information as prescribed by the “Non-In Vitro Diagnostic Device Market Authorization Table of Contents” published by the International Medical Devices Regulators Forum, a group of medical device regulators from around the world that have voluntarily come together to harmonize the regulatory requirements for medical products that vary from country to country.
Nine manufacturers will be recruited to participate in the voluntary pilot. Information on the eligible products and how to apply is available on a new CDRH website.
WHO Announces Mpox as New Name for Monkeypox
“Mpox” (pronounced EM’-pox) is now the preferred term for monkeypox, the World Health Organization (WHO) announced on November 28, 2022. However, both terms will be used simultaneously for one year when the old term for the highly infectious viral disease will be phased out.
The disease was originally named monkeypox in 1970 when the first human case was recorded in the Democratic Republic of Congo (DRC). Since then, most cases have been reported from rural, rainforest regions of the Congo Basin, particularly in the DRC, and human cases have increasingly been reported from across central and west Africa.
“When the outbreak of monkeypox expanded earlier this year, racist and stigmatizing language online, in other settings and in some communities was observed and reported,” the WHO said in explaining the name change. “In several meetings, public and private, a number of individuals and countries raised concerns and asked the WHO to propose a way forward to change the name.”
On the same day as the WHO’s announcement, the United States issued its support for the name change. “We welcome the change by the World Health Organization,” Department of Health and Human Services Secretary Xavier Becerra said. “We must do all we can to break down barriers to public health, and reducing stigma associated with disease is one critical step in our work to end mpox.”
 Mpox is a viral zoonosis (a virus transmitted to humans from animals) with symptoms similar to those seen in the past in smallpox patients, although it is clinically less severe. According to the Centers for Disease Control and Prevention (CDC), mpox was discovered in 1958 when two outbreaks of a poxlike disease occurred in colonies of monkeys kept for research. Despite having originally been named after monkeys, the source of the disease remains unknown. However, African rodents and nonhuman primates (like monkeys) might harbor the virus and infect people, says the CDC.
Assigning names to new diseases is the responsibility of the WHO under the International Classification of Diseases (ICD), through a consultative process which includes WHO member states. Renaming existing diseases is rather rare, though. In accordance with the ICD’s update process, the WHO consulted with global experts, countries, and the public to gather their views and suggestions for the new name. As simple as the new name sounds, it had to undergo a number of rigorous discussions regarding rationale, scientific appropriateness, extent of current usage, pronounceability, usability in different languages, absence of geographical or zoological references, and the ease of retrieval of historical scientific information. The WHO reports that medical, scientific, classification, and statistics experts from government authorities of 45 different countries participated in advisory committees that were heard during the consultation process.
Also discussed was the use of the new name in different languages, concluding that the preferred term of mpox can be used in other languages. Translations, on the other hand, are typically discussed through formal collaboration with relevant government and scientific authorities. In response to the new name, the Associated Press (AP) Stylebook, a prominent writing guide for communicators, was updated online. The AP stated that until the new name becomes more widely known, communicators should use mpox on first reference and mention its former name in one reference later in the story.
US to Create New Global Health and Security Bureau
A virus can spread quickly across borders and around the world, endangering lives, disrupting the everyday functions of countries and communities, and impacting our safety, security, and stability – both in the United States and globally. Recognizing that health security is also national security, the U.S. Department of State plans to create a new bureau focused on global health threats such as COVID-19, Ebola, HIV/AIDS, and other diseases.
In a December 2022 statement, Secretary of State Antony J. Blinken notified Congress of his intention to establish the Bureau of Global Health Security and Diplomacy to ensure that the U.S. government is organized to strengthen global health security and address the growing national security challenges presented by international health crises.
If approved by Congress, the new bureau will combine the efforts of several State Department entities: the Office of International Health and Biodefense (IHB) in the Bureau of Oceans and International Environmental and Scientific Affairs; the functions of the Coordinator for Global COVID-19 Response and Health Security; and the Office of U.S. Global AIDS Coordinator, which oversees the U.S. President’s Emergency Plan for AIDS Relief (PEPFAR).
PEPFAR is considered the largest commitment in history by any nation to address a single disease, prevent millions of HIV infections, save lives, and make progress toward ending the HIV/AIDS pandemic. IHB promotes U.S. national security and economic prosperity by combating biothreats and infectious disease outbreaks through diplomacy.
 “These teams, along with critical partners throughout the government, are already leading our international global health security efforts, and their indispensable functions will continue,” said Blinken. “This new structure would allow our health security experts and diplomats to work more effectively together to prevent, detect, and respond to existing and future health threats.”
Once established, the bureau will be led by Ambassador Dr. John Nkengasong [photo above], currently the U.S. Global AIDS Coordinator, who will then have the title of Assistant Secretary. Born in Cameroon, Nkengasong is the first person of African origin to lead PEPFAR. He previously served from 2017 to 2022 as the first director of the Africa Centres for Disease Control and Prevention (Africa CDC), headquartered in Addis Ababa, Ethiopia. Following his appointment as director, Nkengasong led efforts to create a framework for developing Africa CDC into a full autonomous health agency of the African Union.
Calderón Appointed SENASICA Chief
Mexico’s National Service of Agro-Alimentary Health, Safety, and Quality (SENASICA) appointed Francisco Javier Calderón Elizalde as the new director in chief, this past December. He was sworn in by the Secretary of Agriculture and Rural Development, Victor Villalobos Arambula, to replace Dr. Francisco Javier Trujillo Arriaga. At the swearing-in ceremony, Calderón emphasized the priority of directing SENASICA to facilitate responsible food trade and to seek greater benefits for small-scale producers.
SENSAICA is Mexico’s surveillance, inspectional, and epidemiological arm for agri-food safety. The agency has a strong working relationship with the FDA’s Latin America Office, which has a post in Mexico City.
 Calderón sworn in at SENASICA. Photo courtesy government of Mexico.
Calderón, an agronomist, studied at the Autonomous University of Chapingo and also holds a master’s degree in economics. He has extensive experience in federal public administration, mainly in issues related to the agricultural sector, agricultural economics, public policy, and trade policy. He has diverse experience in multilateral and bilateral negotiations, including with the following agreements:
- Single Free Trade Agreement between Mexico, Guatemala, Honduras, El Salvador, and Nicaragua.
- Mexico-Japan Economic Partnership Agreement.
- Mexico-Peru Agreement.
- Mexico-Colombia Free Trade Agreement expansion.
- Pacific Alliance between Mexico, Chile, Colombia, and Peru.
- Trans-Pacific Partnership Agreement, between 11 countries in the Americas, Asia, and Oceania.
Calderón was also part of the Mexican government team during the negotiation of the U.S.–Mexico–Canada Agreement (USMCA), contributing his expertise regarding market access, trade remedies, labor, environment, and sanitary and phytosanitary measures.
The FDA’s Latin America Office looks forward to continuing ongoing collaborations with SENASICA.
INO Visit to India’s Tata Memorial Center (TMC) Showcasing use of Real-World Data and Innovative Cancer Research
In mid-September, FDA India Office Director Dr. Sarah McMullen and staff visited the Tata Memorial Center (TMC) in Mumbai, one of the oldest and largest cancer centers in India and the world. They met with TMC senior leadership to learn about the center’s research innovations, particularly the use of real-world data for cancer research.
 TMC medical staff (on left) meet with the FDA's India Office staff (on right).
Established in 1941, the TMC remains a major patient care, clinical training, and research institution within India. Research here spans from “bench to bedside.” Their studies include cancer biology laboratory research, as well as large community-based screening trials for common cancers and cohort studies, neoadjuvant and adjuvant treatment, perioperative interventions, surgical trials, and qualitative research to understand the patient journey. The TMC's research goals notably concentrate on patient-relevant endpoints including survival and quality of life measures, needed areas of focus for cancer care in India.
The TMC is using data acquired through the routine care of patients—aka real-world data—to shape and inform some of their research questions and approaches. In particular, the TMC highlighted their use of data from physicians’ off-label uses of medications in cancer care to shape research identifying potentially novel indications that were not a part of the original drug approval. This type of drug-repurposing research is an active area of interest for the cancer research community globally, as well as for the United States.
For example, the TMC highlighted their participation in the “Add-Aspirin” multiregional clinical trial (MRCT) taking place between the United Kingdom, Ireland, and India. This MRCT plans to recruit 11,000 participants to investigate aspirin use after treatment for early-stage solid tumor cancers and any effect on recurrence and long-term survival. As the cost of cancer treatments rise, drug repurposing has opened a window for discovery of new uses for older drugs that could benefit a greater number of patients. Additionally, since this research works with previously approved medicines, efforts taken can reduce the development time and resources typically needed to bring a new chemical entity to market.
The TMC is a major part of India’s National Cancer Grid (NCG), the world’s largest cancer center network with over 150 institutions and partners including patient groups, charitable organizations, and professional societies. The NCG was formed in August 2012 with the directive of linking cancer centers across India. To provide enhanced oversight and compliance of its clinical trials conduct, in 2017 the NCG also established a contract research organization.
As part of the NCG, the TMC provides uniform standards of cancer care and workforce training, and conducts collaborative multidisciplinary cancer research within the country and internationally.
The discussions between the FDA’s India Office (INO) and the TMC during this visit highlighted ongoing applications of real-world data and its potential impact on the standard of care for various cancers in India. Looking ahead, the TMC sees additional potential for collaboration with global researchers to leverage the extensive collection of TMC real-world data and the TMC’s access to India’s National Cancer Grid. The INO looks forward to future interactions to learn more about cancer research at the TMC and in the wider India context, and any implications for the United States.
FDA Reminds Produce Stakeholders of End to Enforcement Discretion for Agricultural Water Requirements
On January 13, the FDA released a fact sheet and constituent update to remind produce stakeholders of the upcoming end of the intended enforcement discretion period for the harvest and post-harvest agricultural water requirements in the Produce Safety Rule for covered produce other than sprouts. This period ends on January 26, 2023, for all farms covered by the rule, other than small and very small businesses. (These terms are defined in the rule.)
As has been done with other Food Safety Modernization Act rules, the FDA plans to take an educational posture for these harvest and post-harvest requirements as the agency begins implementing those requirements. During the first year after the end of the intended enforcement discretion period, the FDA will work closely with international, state, and industry partners to advance training, technical assistance, educational visits, and on-farm readiness reviews to prepare both farmers and regulators for the implementation of these provisions.
Final Guidance Released for the Foreign Supplier Verification Program
On January 10, the FDA issued the final guidance for the Foreign Supplier Verification Program (FSVP) for Importers of Food for Humans and Animals. FSVP is a regulation under the foundational FDA Food Safety Modernization Act that makes importers accountable for verifying foreign suppliers are producing food in a manner that meets U.S. safety standards.
This guidance provides information to importers of human and animal food about how they can comply with the FSVP regulation. It includes recommendations on the requirements to analyze the hazards in food; evaluate a potential foreign supplier’s performance and the risk posed by the food; and determine and conduct appropriate foreign supplier verification activities.
The guidance also addresses how importers can meet modified FSVP requirements in a variety of categories, such as requirements for importers of dietary supplements or very small importers.
The guidance finalizes a draft guidance issued in 2018. In response to comments received to the draft guidance, changes were made to the final guidance, including providing additional clarification regarding to what food the FSVP regulation applies; what information must be included in the FSVP; and who must develop and perform FSVP activities.
Separately, FDA has collaborated with the Food Safety Preventive Controls Alliance to develop training materials to facilitate FSVP compliance by importers.
OGPS Briefs Incoming U.S. Ambassador to the African Union
On January 11, Loyce Pace, HHS Assistant Secretary for Global Affairs, hosted a briefing on the department’s work in Africa for the U.S. Ambassador Nominee to the African Union, Stephanie Sullivan, who was most recently the U.S. Ambassador to the Republic of Ghana.
The African Union is a continental body consisting of the 55 member states that make up the countries of the African continent. The Africa Centres for Disease Control and Prevention, based in Ethiopia, and the newly established African Medicines Agency (AMA), based in Rwanda, are specialized technical institutions of the African Union.
Associate Commissioner for Global Policy and Strategy Mark Abdoo, speaking on behalf of the FDA, provided Ambassador Sullivan with an overview of the FDA’s ongoing work in Africa, including the agency’s efforts to support medical product harmonization and to help build a strong AMA.
 FDA Associate Commissioner for Global Policy and Strategy Mark Abdoo (far left); HHS Assistant Secretary for Global Affairs Loyce Pace (third from left); U.S. Ambassador Nominee to the African Union Stephanie Sullivan (fourth from left).
OGPS Interviews with Global Public Health Leaders in December
In case you missed it, our From a Global Perspective blog featured two interviews with global public health leaders in December.
 Angeli Achrekar (left) and Catherine Hermsen (right).
The first interview was with Angeli Achrekar, Principal Deputy U.S. Global AIDS Coordinator to the U.S. President’s Emergency Plan for AIDS Relief (PEPFAR), to commemorate World AIDS Day on December 1. Dr. Achrekar has served as the second in command to the U.S. Global AIDS Coordinator and supports the daily oversight, coordination, leadership, management, and implementation of PEPFAR to ensure that all its activities and resources — including its annual budget of approximately $6 billion in over 50 countries — are as impactful, effective, and efficient as possible. Early this year she will become the UNAIDS Deputy Executive Director for the Programme Branch, based in Geneva. In her OGPS interview, Dr. Achrekar looked back at her PEPFAR career and talked about the future of the program.
The second interview was with Catherine Hermsen, the FDA Assistant Commissioner for Criminal Investigations overseeing the Office of Criminal Investigations within the FDA's Office of Regulatory Affairs. Hermsen, who retired at the end of December, discussed how her office has adapted to the challenges of globalization and e-commerce to protect public health. “Building viable criminal investigations has become more complicated as globalization and the internet have opened the doors for cases largely centered around bad actors operating out of other countries and illicit supply chains spanning multiple countries. All of these factors have grown increasingly more sophisticated, and, in fact, they have metastasized,” she said.
|
|
|
Back to TOP
 Europe Office Director Ritu Nalubola to Begin New Role as OPLIA Deputy Director
FDA’s Europe Office (EO) Director, Ritu Nalubola, Ph.D., will soon begin a prominent new role at White Oak, as deputy director of the Office of Policy, Legislation, and International Affairs (OPLIA).
In an email to OPLIA staff on December 21, Deputy Commissioner for Policy, Legislation, and International Affairs Andi Lipstein Fristedt said she was “thrilled” to announce that Ritu had accepted an offer to serve as her deputy.
Ritu “will provide executive leadership in the OPLIA Immediate Office (IO) and across OPLIA while leading certain crosscutting initiatives throughout the organization,” Fristedt said. “This will include working with senior leadership to continue to build connections across OPLIA and drive engagement between the IO and all OPLIA offices.”
In addition, Fristedt said that Ritu will “serve a key role in strengthening strategic planning and will be applying her deep policy expertise to continue to drive forward OPLIA’s engagement on a range of important issues, including health equity, high-priority foods policies, and biotechnology.”

Ritu has served as Europe Office Director since July 2018. As an indication of her breadth as director, last year she launched a new FDA initiative in Europe, working with the FDA’s Office of Criminal Investigations and in partnership with the Paris-based Organisation for Economic Co-Operation and Development (OECD), to combat the global threat of illicit trade in health products. The initiative featured a series of three meetings to advance a whole-of-governments approach to the problem and securing State Department support for a new FDA posting to the U.S. Mission to the EU in Brussels (to be filled by an OCI special agent) and new office space for expansion.
And just last week she began to implement a forward-leaning vision for U.S.-EU collaboration to achieve a faster and more effective response to global food outbreaks — by hosting the first U.S.-EU dialogue on whole genome sequencing in outbreak response and pathogen surveillance. While Ritu has helmed the EO, the office has significantly expanded its foods portfolio. She began this effort first by understanding the challenges, sensitivities, and priorities surrounding the FDA’s various centers and offices overseeing food issues before charting the EO’s own contributions. She and her team facilitated the resumption of U.S.-EU trade in bivalve molluscan shellfish following the FDA’s first U.S.-EU shellfish equivalence determination, coordinated establishment of the U.S.-EU seeds-for-sprouting export certification program in collaboration with the U.S. Department of Agriculture, and renewed efforts as the FDA began to move into the next phase of systems recognition discussions with the European Commission (EC).
|
 Europe Office group members, as staffed during the height of the pandemic years. Ritu Nalubola (seated front right, in blue).
“It has been a great honor to represent the FDA and serve under the U.S. Mission to the European Union,” said Ritu. “Throughout my tenure, I have endeavored to represent the FDA with professionalism, competence, integrity, and vigor. During the remainder of my term, I intend to continue to work to align the office with OGPS’ strategic plan and position the office for a successful future.”
As her tenure coincided with the COVID-19 pandemic, Ritu successfully diverted resources to address COVID-related issues. In collaboration with FDA, European, and U.S. government stakeholders, she organized and hosted the first global COVID-19 vaccines regulators workshop to discuss strategies and facilitate the development of SARS-CoV-2 vaccines and promote regulatory convergence. And throughout the pandemic, she directed her staff as they assisted FDA headquarters, the U.S. Mission to the EU, and EU partners on numerous COVID-related issues, including exchanges of inspection reports, regulatory authorizations, supply chain disruptions of critical medical products, export controls, shipments of specific vaccines and drug substances, fraudulent products, vaccination certification schemes, and related national testing programs.
Similarly, Ritu and her EO colleagues made inroads in developing competencies in medical devices and building trust with both the FDA’s Center for Devices and Radiological Health (CDRH) and the EC, as well as building relationships with member states’ competent (regulatory) authorities to gather information related to postmarket issues. These gains have resulted in routine interactions and more positive working relations between the FDA and the EU, a long-sought goal for the Europe Office.
One of Ritu’s significant achievements has been to foster global regulatory cooperation in niche areas by leveraging U.S. and EU expertise and leadership. Applying her keen interest in emerging technologies, she identified the need for early scientific and regulatory exchanges to promote policy convergence and, ultimately, facilitate innovation. She established the global regulators biotechnology forum, leading and hosting three global workshops on the use of genome editing in plants and animals — a unique forum allowing for open, informal discussions among regulators on complex, sensitive issues related to agricultural biotechnology. She and her team also initiated a think tank comprising a core group of global regulators focused on digital health technologies to support the FDA’s objectives at the broader international forum.
Perhaps Ritu’s most salient accomplishment during her tenure was transforming the Europe Office into a cohesive unit with a stronger organizational identity — both within and outside the FDA — while solidifying the agency’s presence in Europe. She restructured the office with additional U.S. direct and locally employed staff to better position OGPS to meet the FDA’s strategic interests in the region. Under her leadership, the EO staff put considerable effort into establishing the office as a key FDA agent for information gathering and analysis, strategic leadership, and engagement with European counterparts. Now, in addition to the EO’s existing thriving relationships with EU entities — including the EC and the European Medicines Agency — the office has a robust network of contacts at key member state agencies, an invaluable tool for gathering timely information on both rapidly evolving concerns and routine policy questions.
Over the course of her public service career, Ritu has advised senior leadership at the FDA, the Department of Health and Human Services, and other U.S. government agencies on complex and crosscutting policy issues, including biotechnology, nanotechnology, food safety, nutrition, and trade-related matters. Prior to joining the Europe Office, she spent seven years in the FDA’s Office of Policy in the Office of the Commissioner as a senior policy advisor on emerging science policy issues, with a focus on sound policy approaches against the backdrop of scientific uncertainty inherent to novel technologies. Before starting her career at FDA in 2000, Ritu worked with the U.S. Agency for International Development on international public health issues.
“I had the good fortune of spending time with Ritu in Europe this past fall,” said Fristedt. “I am confident that Ritu’s leadership skills, collegiality, and substantive expertise, combined with her intimate familiarity with FDA, HHS, and U.S. government broadly, will be a major asset to our office.”
Over the next few months, Ritu will start working with OPLIA IO as she continues to wrap up her tenure in Brussels.
Natalie Mickelsen Departs India for the Land of the Rising Sun

The India Office’s (INO) Natalie Mickelsen finished her 4-year foreign tour in December after advancing from consumer safety officer to supervisor and finally to deputy director. An accomplished veterinarian, Natalie will continue her overseas adventures by joining the U.S. Department of Agriculture’s Animal and Plant Health Inspection Service (USDA-APHIS) in Japan as a veterinary medical officer focusing on epidemiology.
As OGPS’ only veterinarian on staff, Natalie will be missed for her thoughtful and perceptive insights, and her skill in working across agencies and integrating multiple perspectives.
Serving in the FDA’s Office in New Delhi during the COVID-19 pandemic created a number of unique challenges for the INO staff, and Natalie was instrumental in helping the staff to overcome them. She created a procedure for resuming in-person inspections in India, making it possible for consumer safety officers to conduct mission-critical inspections, even while navigating travel restrictions and other logistical difficulties. Her procedure was so effective that other foreign regulatory authorities emulated the approach in developing their own procedures.
|
 Additionally, during the height of the pandemic, the INO continued to support investigational activities of bilateral importance, in particular, responding to a salmonella outbreak in the United States that was attributed to shrimp from India. Natalie and her team worked with their Indian regulatory counterparts to facilitate their sharing with the FDA of the initial findings of the government of India's investigation of the manufacturing facility. Once COVID-19 conditions improved, the INO team followed up on problematic conditions at the facility by conducting their own FDA inspection. The team is now drafting a technical article on the insights learned from the investigation, for later journal publication.
During Natalie’s INO tenure, she was able to visit institutions and meet officials from India’s regulatory counterparts and public health organizations across the country. Highlights included: interacting with officials on topics of Ayurveda education and research while visiting the Gujarat State Model Institute for Ayurveda Sciences at Gandhinagar and associated Ayurveda Hospital; contributing to the annual regulatory forums of Indian state counterparts and the Central Drugs Standard Control Organisation; and speaking at the Good Aquaculture Practices workshop with the Marine Products Export Development Authority in Kochi and Seafood HACCP trainings with the Export Inspection Council in Mumbai.
 Supporting the FDA’s mission as a part of the U.S. Embassy in New Delhi allowed Natalie to work closely with interagency colleagues from the Department of Health and Human Services, Centers for Disease Control and Prevention, U.S. Department of Agriculture, and Department of State. “I found my love for working overseas and conducting diverse, interagency work,” she said. “I love that no two days are the same. I’ve learned the importance of diplomacy and relationships inside and outside of our agency and country. The biggest lesson learned is the importance of a great team. Our office works hard, and cares about each other, which makes leaving so difficult! I love these guys.”
Starting this month and for the next 10 months, she’ll be in the D.C. area for Japanese language studies provided by the State Department before deploying to Tokyo in late 2023.
Europe Office’s Louati Receives Prestigious Award
International Policy Analyst Claudia Louati in the FDA’s Europe Office (EO) received the State Department’s Mission Honor Award at a ceremony on December 12, 2022. The U.S. Mission to the European Union’s (USEU) Chargé d'affaires, Kelly Adams-Smith, presented the award to Louati for “outstanding leadership and invaluable contributions to the FDA’s whole-of-governments approach to combatting illicit trade in health products.”
 Claudia Louati (left) and Kelly Adams-Smith (right).
During 2022, the EO, in close coordination with the FDA’s Office of Criminal Investigations (OCI), embarked on a new initiative to address the growing public health threat posed by the transshipment of illegal medicines and medical devices that originate from a third country and transit through Europe, usually without entering customs, before shipment to the United States. This serious problem became more urgent during the pandemic as e-commerce proliferated and criminal networks took advantage of product shortages and patients’ vulnerability.
Working with OCI, the EO initiated and executed a series of activities to promote a whole-of-governments approach with key stakeholders in the region. These activities included events, large and small bilateral and multilateral meetings, and making connections with national, regional, and global organizations that play a role in health product trade, including those involved with customs and border protection, taxation, patent and trademarks protection, and drug regulation. It was Louati who stepped up to take on the additional role of supporting the EO’s work on this new initiative to help ensure its success.
With Louati’s coordination, the EO and OCI were able to conduct over 20 visits and meetings with a broad range of organizations, including Interpol, Europol, Organisation for Economic Co-operation and Development (OECD), Universal Postal Union, World Health Organization, World Customs Organization, EU’s anti-fraud office, DG SANTE, as well as regulatory counterparts in Germany, France, and Switzerland. It was her diligent planning and tactful outreach to these diverse stakeholders — many of whom were not in the office’s routine regulatory community network — that allowed the EO to create strong momentum for implementing the FDA’s vision for a whole-of-governments approach with European and global colleagues.
As a result, the EO was able to conduct three large global workshops in partnership with the OECD. The first two, conducted virtually in May and July 2022, included nearly 70 attendees from 18 countries. The final event was an in-person, two-day workshop in September 2022, with about 110 participants from 20 countries held at the OECD campus in Paris. As this was a high-profile public health issue, the FDA’s senior-most leadership (FDA’s Deputy Commissioner, Associate Commissioner, and Assistant Commissioner) traveled to Brussels and Paris for the September in-person event. Louati either led, coordinated, or was an integral part of the EO’s complex and involved preparations for each event, as well as planning the itinerary, meeting opportunities, and background briefings for the visiting FDA leadership. Her total dedication, substantive policy expertise, meticulous planning, and attention to detail are the reasons agency efforts have been such a great success.
The value of these efforts is reflected in the strong interest expressed by participants of the workshops to continue the forum, develop a concrete set of workstreams, and reconvene in one year. Louati is continuing to work on these follow-up items and support the EO’s future work on combating illicit trade. Successful planning, development, and implementation of this initiative is an example of how the FDA/USEU is providing global leadership to advance international diplomacy and strategic engagement on a topic of high public health significance. EO Director Ritu Nalubola noted that, “The success of this initiative could not have been possible without Louati’s detailed policy analysis, exceptional dedication, and overall outstanding performance.”
This is the second award during the past year given to the EO’s locally employed staff. In June 2022, International Policy Advisor Alessandro Fiorelli was awarded the U.S.-EU Meritorious Honor Award in recognition of his “exemplary and outstanding contributions enabling the resumption of trade in shellfish between the U.S. and the EU, ending a decade-long ban in transatlantic commerce.”
|
|
|
Back to TOP
  Left to right: Constance Richard-Math, Geurlain Ulysse, Eric Reisenauer, Danijela Stojanovic.
Incoming
Constance (Connie) Richard-Math
Connie Richard-Math joined the India Office in December, taking on the challenge of being the lone supervisory consumer safety officer — and responsible for covering all product areas. Richard-Math joined the FDA in 1998 and has supervisory experience with multiple FDA offices, including as the director of the Investigations Branch at the Office of Regulatory Affair’s (ORA’s) district office in Baltimore. She has broad experience in both domestic and import regulatory activities, having managed enforcement under all of the FDA’s program areas. She also has external experience as a consultant regarding medical products. Richard-Math is a graduate of Iowa State University with a Bachelor of Science in animal science.
Guerlain Ulysse
Guerlain Ulysse joined the India Office in December as a consumer safety officer specializing in drug products. Since joining the FDA in 2015, he has held the role of consumer safety officer for the ORA’s New Jersey District Office and drug specialist for the ORA’s Office of Pharmaceutical Quality Operations, Pharma Division I. He has also completed details as a supervisory consumer safety officer and has been a part of ORA’s Dedicated Foreign Drug Cadre, conducting various international drug inspections. Ulysse holds a master’s degree in biotechnology from the University of Maryland Global Campus and a bachelor’s degree from the University of Michigan. He has coauthored scientific publications in the field of biomedicine and biotechnology.
Eric Reisenauer
Eric Reisenauer joined OGPS in October 2022 and deployed to the China Office in early January, as a consumer safety officer (CSO) for human and animal foods. Reisenauer has been with the FDA since 2009, serving as a CSO at the ORA’s New York District Office. His most memorable regulatory action was obtaining a warrant to seize the contents of a bakery for filth conditions and working with the U.S. Marshals to execute the warrant. Reisenauer was also a member of the ORA’s Foreign Food Cadre from January 2016 to August 2020, doing inspections in Europe, Asia and the Pacific, and Latin America. He is a graduate of SUNY Geneseo with a Bachelor of Science in biochemistry.
Lateral
Danijela Stojanovic
Danijela Stojanovic has permanently joined the Europe Office as an international policy analyst focusing on medical products; she is based stateside at White Oak. This follows on the heels of her detail in this position during the second half of 2022. She continues her work on the Parallel Scientific Advice program and supporting OGPS’s work with international clusters and with the European Medicines Agency-FDA Liaison program. Stojanovic leaves her epidemiologist position with CDER’s Office of Surveillance and Epidemiology to become part of our OGPS family. She is an officer in the U.S. Public Health Service Commissioned Corps.
|
|
|
Back to TOP
 Recent communications from OGPS to our international stakeholders (list does not include twice-weekly FDA Roundup summaries), November 3, 2022, through January 13, 2023.
|
|
|
Back to TOP

|
January 20
|
World Neglected Tropical Diseases Day
|
|
January 26
|
VRBPAC meeting – future vaccination regimens for COVID-19
|
|
February 4
|
World Cancer Day
|
|
February 13-16
|
Global Harmonization Working Party, Riyadh
|
|
February 16-18
|
World Spice Congress, Mumbai, India
|
|
February 24-26
|
BioAsia 2023 Annual Conference, Hyderabad, India
|
|
|
Read thought-provoking pieces covering international topics in
From A Global Perspective. |
|
Please follow @FDA_Global. |
|
|
Subscribe to Global Update
Don't miss out on future issues of Global Update or other international news from OGPS. To sign up, click "Manage Subscriptions" in the below footer then follow the prompts and select the "International Programs" box.
|
|
|
|
|



