|
 
April 2024
Volume 24, Issue 4
The U.S. Food and Drug Administration’s (FDA) MedSun program provides this monthly newsletter to inform patients and patient advocates about FDA information on medical device related topics. The MedSun program, launched in 2002 by the FDA’s Center for Devices and Radiological Health (CDRH), uses a secure online reporting system to receive medical device adverse event reports from a network of over 300 clinical facilities across the United States. MedSun sites work collaboratively with the FDA to assist in detecting, understanding, and sharing information concerning the safety of medical products and play a critical role in the FDA’s postmarket surveillance efforts.
|
|
|
The reports that follow represent a cross section of device related events sent by MedSun Representatives during the prior month. The reports are presented as submitted by MedSun Representatives and in some instances, have been summarized and edited for clarity.
|
|
Type: Catheter, Intravascular, Therapeutic, Short-term Less Than 30 Days
Manufacturer: Teleflex Inc.
Brand: Pressure Injectable Arrowg+ard Blue Plus Two-Lumen CVC
Cat: ASK-46702-PWBH1
Event: After a surgical procedure for the insertion of a double lumen Arrow Central Venous Catheter (CVC), medical imaging identified a possible retained foreign object. A computed tomography scan (CT) was performed, which confirmed a retained foreign object in the right atrium. The patient was moved from ICU to Interventional Radiology (IR) to retrieve the foreign object. In IR, the CVC was removed, and the foreign body was no longer observed in medical images. The suspected foreign object was the distal end of the CVC. Teleflex was notified and a physician suspected that the perceived foreign object observed on the CT was in fact the plug of the catheter, a feature of all multi-lumen catheters which separates the lumens, allowing for independent infusions. The concern is that this plug was radiopaque and mistaken for a retained foreign body, resulting in a critically ill patient being moved from the ICU to IR for a potential retrieval procedure. Teleflex was engaged in determining the issue. Teleflex acknowledged sporadic reports of this issue and similar CVC's being misidentified as retained foreign bodies in medical images.
Type: Set, Tubing, Blood, With and Without Anti-Regurgitation Valve
Manufacturer: Outset Medical, Inc.
Brand: Tablo(R) Cartridge
UDI-DI: 00850001011273
Model: PN-0008544
Cat: PN-0004220
Lot: M23137L02S01, M23156L03S01 (x2), M23166L02S01 (x3), M23166L03S01
Event: This is a summary of multiple reports: These medical device reports highlight several recurring issues with the Tablo dialysis cartridges, including leaking blood and saline from various connection points, blood lines improperly seating, and air bubbles within the Tablo Hemodialysis System. In some cases, we inspected the cartridge, and found the connection to the loop of tubing that goes around the blood pump had leaked and dripped onto the lower connection and then out the bottom of the cartridge. These problems have led to blood loss, increased potential contamination risks, and interruptions in dialysis treatment. Currently, there are no adverse patient outcomes.
|
|
Neonatal and Pediatric Reports |
|
 |
|
These reports describe medical device events involving neonatal or pediatric patients, or those events involving a medical device that is indicated for use in neonatal and pediatric patient populations.
The FDA defines pediatric patients as those who are 21 years of age or younger (that is, from birth through the twenty-first year of life, up to but not including the twenty-second birthday) at the time of the diagnosis or treatment.
|
Type: Electrode, electrocardiograph
Manufacturer: Conmed Corp.
Brand: Softrace
UDI-DI: 30653405038973
Model: 2310-003
Lot: 202303105 (x2)
Cat: 2310-003
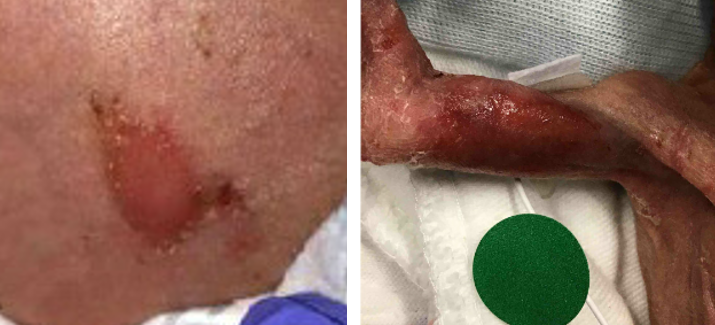
Event: This is a summary of multiple reports: The facility sent multiple MedSun medical device reports detailing adverse events with ConMed Softrace EKG electrodes. The reports detailed skin burns, wounds, and marks linked to the electrodes. One report indicates a skin burn on the right upper arm. The gel that was underneath the lead was swollen and falling off. The burn had taken off the first layer of skin, and the arm was bleeding. Another report describes a wound on the abdomen caused by limb EKG lead electrodes, requiring consultation with a wound team and additional patient treatment. The third report involves a patient with sensitive skin developing marks from the leads on the chest and abdomen, which were discovered and documented by a nurse during a subsequent shift.
Left photo: Abdominal burn/wound; Right photo: Underside of left upper arm showing burn/redness.

Type: Set, Administration, Intravascular
Manufacturer: Hummingbird Med Devices Inc. and Nexus Medical Inc.
Brand: Split Septum Micro T-Connector and IV Plus
UDI-DI: 00850584007014
Model: NMT8046
Cat: NMT8046
Photo: Crack shown on Hummi T-connector.
|
Event: This is a summary of multiple reports. This facility received multiple reports about problems with this device and believes together, they show a device failure pattern, raising concerns about reliability and safety in critical care settings. The Hummi Micro-Draw Blood Device was leaking. A nurse observed the monitor alarming and saw the arterial line tracing was dampened with negative numbers. Staff discovered the Hummi T-connector was cracked and there was blood loss from the arterial line site. The nurse applied pressure and medical staff changed the T-connector piece as well as the safe set and re-dressed the site. The leaking Hummi device may have contributed to a RBC blood transfusion the patient later needed.

Type: Set, Administration, Intravascular
Manufacturer: Hummingbird Med Devices Inc. and Nexus Medical Inc.
Brand: Split Septum Micro T-Connector and IV Plus
UDI-DI: 10815879021424
Model: NMT8046
Lot: 23110
Cat: NMT8046
Photo: Cracked and separated Nexus IV Plus T-connector.
|
Event: This is a summary of multiple reports. This facility received multiple reports about problems with this device and believes together, they show a device failure pattern, raising concerns about reliability and safety in critical care settings. The patient had a 40 point drop in systolic blood pressure with a dampened waveform. Staff discovered blood loss from a cracked T-connector, which was temporarily managed by applying pressure and taping the crack and then replaced under medical supervision. There was minimal blood loss.
|
|
FDA Seeks Comments on Informed Consent Guidance Document
To improve the informed consent process for potential research participants, the FDA and the Office for Human Research Protections have published a draft guidance titled “Key Information and Facilitating Understanding in Informed Consent.” The FDA requests public comment on the guidance by April 30, 2024. This draft guidance provides recommendations on how to implement current requirements under the revised Common Rule, including that:
- Informed consent begins with key information about the research presented in a clear and concise manner.
- Informed consent is presented in a way that facilitates understanding of the reasons why someone might or might not want to participate in the research.
Select Updates to the Premarket Cybersecurity Guidance

On March 13, 2024, the FDA issued the draft guidance “Select Updates for the Premarket Cybersecurity Guidance: Section 524B of the FD&C Act” which proposes select updates to the final guidance “Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions.” This draft guidance, when finalized, will identify the information FDA generally considers necessary for cyber devices to support obligations under the new amendments to the Federal Food, Drug, and Cosmetic Act (FD&C Act) for ensuring cybersecurity of devices that was implemented on March 29, 2023.
The select update discusses who is required to comply with section 524B, devices subject to section 524B, and documentation recommendations for applicable premarket submissions. For example, the select update discusses that coordinated vulnerability disclosure (CVD) and related procedures should include coordinated disclosure of vulnerabilities and updates identified by external entities and by manufacturers of cyber devices, and manufacturers should have procedures to carry out such disclosures.
The FDA will host a public virtual webinar on April 30, 2024, to discuss this draft guidance. Suggestions and comments regarding the draft guidance can be submitted to the public docket by May 13, 2024.
See FDA’s medical device Cybersecurity website for additional information and resources.
|
|
|
Links to FDA CDRH Databases and Other Information Sources |
|
U.S. Food and Drug Administration
10903 New Hampshire Ave.
Silver Spring, MD 20993 |
|
|
|
|