| |
|
If your email program has trouble displaying this email or images, view as a webpage.
|
|
|
Back to TOP
 
John Weiner Joins OGPS as Office of Global Operations Director
John “Barr” Weiner, J.D., is the new director of the Office of Global Operations (OGO), one of the three suboffices in the FDA’s Office of Global Policy and Strategy (OGPS).
In his new role, Weiner will oversee the FDA’s foreign offices located in strategic locations throughout the globe, as well as international policy advisors, experts at FDA headquarters who are responsible for bilateral and regional engagement, and global operations and program support staff. He takes over from Mark Abdoo, associate commissioner for global policy and strategy, who was serving in that position on an acting basis, in addition to his other duties.
OGO is an integral OGPS suboffice. It strives to:
- Increase the FDA’s knowledge of the countries and regions from which FDA-regulated products are imported and where other FDA-regulated activities occur, enabling the agency to make more informed decisions and more efficiently allocate its resources.
- Expand foreign government and industry awareness and understanding of FDA regulations, furthering regulatory coherence and compliance.
- Foster international regulatory convergence and harmonization through bilateral and regional engagement to enhance regulatory effectiveness and efficiency and opportunities for coordination and collaboration.
- Conduct in-country/in-region inspections in cooperation with the FDA's larger foreign inspection program.
“OGO already does so much, and has the potential to do even more, to help the FDA pursue robust, coherent bilateral and regional engagement, as part of OGPS’ overarching efforts, to increase regulatory efficiency and effectiveness globally,” Weiner said. “In this way, we can enable access to safe and effective medical products and safe foods and other consumer goods for the American public.”
Immediately prior to his appointment to OGO director, Weiner was on loan to OGPS to advise Associate Commissioner Abdoo on certain policy matters across the medical products space, including supply chain resilience and long-term international strategic planning for the agency. He will continue to support OGPS policy development in his new role.
Weiner comes to OGPS from the Office of Combination Products (OCP) where he served as the associate director for policy. In his time at OCP, Weiner was a principal architect of the FDA’s combination products regulatory program — leveraging the regulatory programs for drugs, devices, and biological products and ensuring appropriate coordination across the FDA throughout the regulatory life cycle from investigation to premarket review, to marketing, manufacture, and postmarket change management. His work has involved engagement with leadership and subject matter experts across the FDA, collaboration with foreign counterparts, standard-setting bodies, regulated entities, trade associations, and other interested parties.
Earlier, he was an associate chief counsel for the FDA, focusing on drug regulation and related innovation and competition issues, and on crosscutting topics including the use of nanotechnology — briefly serving as the FDA’s policy lead for that issue. In addition, he served as the FDA’s liaison to the Office of the United States Trade Representative and other components of the U.S. government on issues relating to pharmaceutical trade and competition, while participating in multiple rounds of bilateral and regional trade negotiations as a technical advisor.
Before coming to the FDA, Weiner was in private practice, focusing on food and drug, environmental, and related public international and trade law, providing regulatory counsel and litigation strategy. He represented clients before regulatory agencies, Congress, and regional and multilateral organizations — and in negotiations of international agreements, including the Kyoto Protocol on climate change, the Stockholm Convention on persistent organic pollutants, and the Cartagena Protocol on biosafety.
Weiner received a Bachelor of Arts degree from Princeton University and his Juris Doctor degree with honors from the Columbia University School of Law.
McMullen to Make Leap from India to China
The FDA’s India Office (INO) will soon be losing Director Sarah McMullen, Ph.D. — to the China Office! She’s been at INO over five years, and since overseas staff are limited to six years in country per the policy of the Department of Health and Human Services, this summer she’s moving to the director role in Beijing.
McMullen began her India tour as a supervisory consumer safety officer, overseeing the inspectional activities of INO-based investigators across foods, clinical trials, and pharmaceuticals. As leadership positions opened within this OGPS-managed foreign office, she applied and was selected for deputy director, then director, and has now been in that role for three years. “Having served in several positions with the office has been incredibly helpful in understanding the operations and the needs in each area,” McMullen said.

Dr. Sarah McMullen, director of the FDA’s India Office. (Artwork incorporating a portrait courtesy NDTV/Profit and drug background from Getty Images)
Prior to joining OGPS, McMullen, who has a doctorate in inorganic chemistry, spent nearly 15 years with the FDA’s Office of Regulatory Affairs (ORA) working as a chemist, tissue residue specialist, consumer safety officer, and manager. While there, she performed inspections across FDA-regulated commodities, and her analytical efforts included pharmaceutical quality testing and analysis of contaminants in seafood, agricultural, and tobacco products. The last several years of her ORA experience focused on import operations.
McMullen assumed her leadership roles in INO during the COVID pandemic. It was an intensely challenging time, both personally and professionally. India responded to the pandemic by suddenly imposing export restrictions on medical products. With so many products sourced from India, it was a blow to the United States. INO and other U.S. Mission agencies stationed at the U.S. embassy in New Delhi worked with industry to show the government of India that the country produced enough capacity that it could support both domestic and export markets. “We were able to help diplomatically to ensure the flow of essential medicines to the U.S. when they would otherwise have been restricted from export,” said McMullen, about this first wave of COVID in India.
India was then hit much harder during what is called the second (Delta variant) wave of the pandemic. This time, the United States sent medical products to India. INO chaired the Mission’s Interagency Specification Task Force that helped to ensure that regulatory requirements were met for the incoming medical products. The office’s many efforts during that time earned the staff multiple awards from the U.S. State Department. McMullen described that period in the article, Doing the Needful, published in OGPS’ From a Global Perspective blog series.
The medical products space — including vaccines, clinical trials, pharmacovigilance, and substandard and falsified products — has been an important focus of her office’s engagement work with Indian industry and regulators.
A primary emphasis has been campaigning for higher manufacturing quality standards and urging both state and national regulatory authorities to devote more resources to regulatory oversight. Notably, last year, the FDA co-hosted an “indigenous” summit on quality with industry. Concurrently, and in response to several high-profile lapses in quality, the government of India hosted a series of internal meetings to deliberate on how to ensure more robust and consistent oversight for pharmaceutical manufacturing. In addition, her office has sponsored joint regulatory forums with four state regulators, thus enabling them to observe FDA inspections.
McMullen's efforts to combat illicit and falsified products included working with John Verbeten, the deputy director for enforcement and import operations with the FDA’s Office of Regulatory Affairs. Together they conceived of a multi-agency operation, Operation Broader Sword, working closely with the U.S. Customs Attaché at Embassy New Delhi.
During June 2023, Operation Broader Sword targeted illicit medical products shipped from India to the United States using the international mail system. In collaboration with the government of India’s Directorate of Revenue Intelligence, this bilateral operation stopped over 500 shipments and provided both U.S. and Indian law enforcement a better understanding of international criminal networks — knowledge that will further advance the regulatory efforts of both countries.
“Being able to leverage in-country U.S. expertise and collaborative relationships among several stakeholders led to the seamless planning and execution of this extensive operation,” said McMullen.
Under McMullen’s leadership, the INO also sought to better understand the landscape, operations, and concerns of two ancient commodity sectors that market to the United States — spices, which thrive in India’s lush geography and climate, and herbal formulas that are commonly used in traditional or Ayurvedic medicine. Her office has met with Indian regulatory counterparts, attended related conferences, and toured a variety of established growing, processing, academic, and medical facilities, observing not only in a learning mode but also communicating and advocating for the FDA’s regulatory expectations.
“The demand for traditional medicine products is ever-increasing in the U.S. Working at the source locations such as India and China to increase product compliance will not only benefit consumers but also allow for more efficient use of the FDA’s resources in import oversight,” McMullen said.

Last June, INO facilitated regional training in the Indian states of Kerala and Assam (significant centers of spice commerce) on Good Agricultural Practices and Good Manufacturing Practices (GMP), targeting the unique needs of growers and processors of spices, herbs, and medicinal plants. This was the first time the FDA’s GMP training in India was specifically focused on the unique needs of the processors of these botanical crops — an important emphasis, since Indian-grown spices and herbs often end up in the U.S. marketplace as FDA-regulated culinary seasonings and dietary supplements.
McMullen will begin her director role at the FDA China Office in late summer. “India has taught me so much and I will forever be grateful for the time my family and I were able to spend here,” she said. “I am grateful for each member of our INO family, their presence and engagement are so valuable in facilitating the ongoing efforts in India. While I am sad that this chapter is coming to a close, I’m excited to join another amazing office in China. As a lifelong learner, I’m thrilled and daunted by all I have to learn in a new country, a new context, and new work, but I know I’ll have amazing people next to me on the journey.”
|
|
 
This 2024 International Women's Day, OGPS celebrated the women in our Office of Trade and Global Partnerships (OTGP) in our annual social media post in observance of the Day. This office serves as the FDA lead for addressing issues related to international trade of regulated products, mutual recognition agreements, and entering into arrangements and sharing information with global counterparts. Read more about OTGP by clicking on the image below.

For the for the third year in a row and consistent with Women's History Month, the FDA fielded an all-woman panel for its DIA Europe Town Hall session, held on March 13. The topics discussed during the interactive and lively session ranged from early interactions on complex products to all the different ways the FDA can collaborate with its European regulatory partners.

The speakers included (from left to right) Katherine Tyner, the FDA's liaison at the European Medicines Agency in Amsterdam, and members of the FDA's Europe office in Brussels — Katie Serrano, director; Shannon Thor, deputy director; and Ioana Ulea, policy advisor.
The day before at DIA Europe, Shannon Thor participated in a panel discussion on combination products, where she stressed the importance of early engagement between regulators and improving coordination, collaboration, and communication to support comprehensive risk management.
|
|
|
Back to TOP

Million Dollar Request to Expand the FDA’s Foreign Footprint
Funding to increase the scope of the FDA’s foreign work was included in the $7.2 billion requested by the FDA in the President’s proposed fiscal year 2025 budget unveiled on March 11.
The FDA is seeking $1 million “to support the expansion of the agency’s foreign-office footprint,” increase agency resources to “facilitate timely inspections of foreign facilities in specific countries,” and “strengthen imported products oversight,” the agency said in a same-day press release.
The $1 million would “begin the process of expanding our foreign footprint,” Mark Abdoo, associate commissioner for global policy and strategy, said in remarks at the International Medical Device Regulators Forum on March 12. “This ultimately will include reimagined and expanded offices in India and Latin America, an Asia regional office, a presence in Africa, and changes to our Europe office to better manage the wealth of clinical trials that are conducted there,” he said.
The FDA recently marked the 15th anniversary of the opening of its first foreign office, in Beijing. “The FDA foreign offices were initially conceived to manage the issues the FDA was facing at the time — a boom in outsourcing of medical product and other manufacturing to India and China and an increasing reliance on produce imported from Latin America,” Abdoo said. “While these factors continue, there are new challenges emerging that require an expanded FDA foreign presence for us to meet the challenges of the next 15 years.”
Congress is already expressing interest in having the FDA devote more efforts internationally. The agency’s fiscal 2024 funding package, enacted earlier this month, contained language directing the FDA to develop a plan within the next 90 days in cooperation with the State Department for expanding the agency’s international presence.
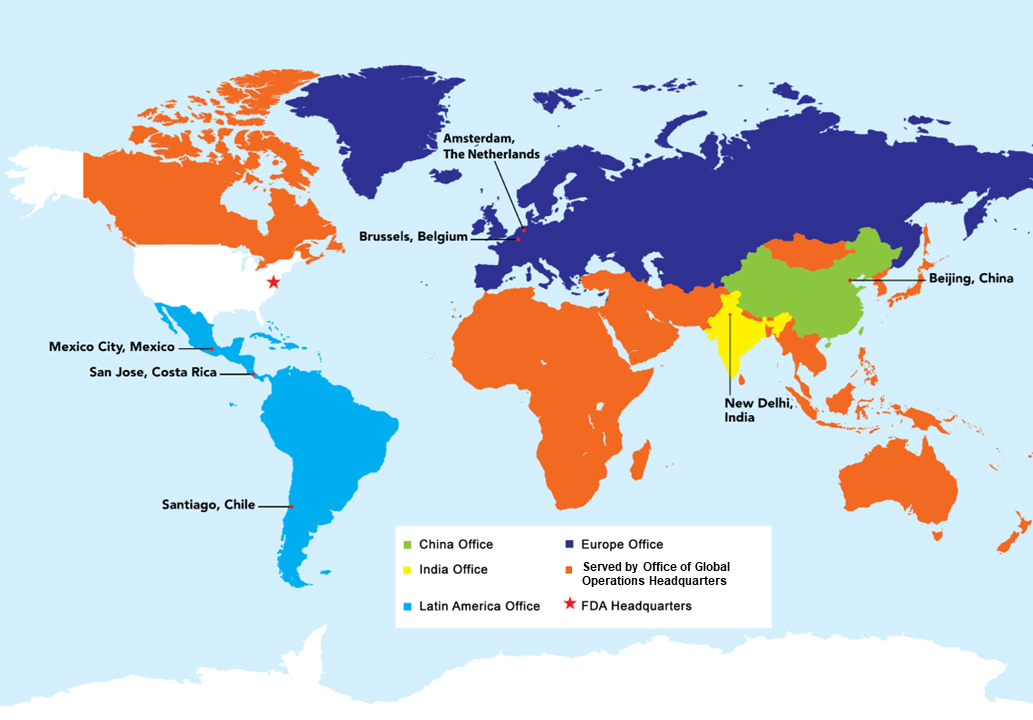
Current regions covered by the FDA's foreign offices and OGPS' Office of Global Operations' headquarters staff.
Reliance: A High Priority at IMDRF Meeting
Reliance — where regulators make decisions in part by taking into account or giving significant weight to the findings of another institution — took center stage at the 25th Session of the International Medical Device Regulators Forum (IMDRF) held in Washington D.C. March 11-15.
The IMDRF is a voluntary group of medical device regulators from around the world whose aim is to harmonize the world’s approach to the regulation of medical devices.

The FDA’s Center for Devices and Radiological Health Director and IMDRF Chair, Jeff Shuren, M.D., J.D., said, “one of the most effective ways to harmonize is through reliance,” making it the focal topic for the daylong workshop that kicked off the event.
“By building trust through the sharing and protection of information and expertise, regulators may rely on the pre- and post-market decisions of other jurisdictions, which can save significant time and resources. Reliance can also increase market and patient access to the best devices,” said Dr. Shuren. “We view models of reliance as a win for all of public health.”
Throughout day one, speakers from both regulatory agencies and industry discussed what is reliance and why it’s importance, the role of standards and the World Health Organization (WHO), and the use of reliance in both the pre-market and post-market setting.
“Many countries have limited regulatory capacities but still need to facilitate access to quality-assured medical products said Hiiti Sillo, Unit Head, Regulation and Safety, at the WHO. “Reliance promotes better use of limited resources and strengthens global regulatory oversight,” he said.
Participants agreed that for reliance to advance requires transparency, trust, and IMDRF support.
In 2011, when the IMDRF was stood up in place of its predecessor organization, the Global Harmonization Task Force, there were seven member nations and one official observer, the WHO. Today there are 11 member nations, two other official observers, Argentina and Switzerland, seven associate members and four regional harmonization initiatives which it supports. But those numbers don’t reflect the much broader interest in the topic of harmonization, as marked by the event’s record turnout of 1,200 people, representing more than 60 countries and 550 public and private sector organizations.

FDA Associate Commissioner for Global Policy and Strategy Mark Abdoo speaking at IMDRF session about the global efforts underway in his office.
While day one of the public-facing portion of the meeting focused on reliance, day two was given over to regulatory updates by the 11 member states and the three official observers, with opening remarks by FDA Associate Commissioner for Global Policy and Strategy Mark Abdoo who outlined a few of the global efforts underway in his office. Presentations from both days have been posted on the IMDRF website.
One of the core principles of the IMDRF is that its work should be available to all, Shuren said. IMDRF working groups develop technical guidances and other documents to provide policies and best practices for the entire medical device community. This information can be quite technical, and so IMDRF members are considering how best to provide training on these documents.
The FDA will host a second IMDRF session in the United States in September, with a location yet to be determined.
Project Orbis Continues to Expand
Nineteen unique oncology drugs were approved by the FDA and its international partners under the Project Orbis framework in 2023.
Project Orbis provides for concurrent submission and review of oncology products that can result in simultaneous decisions by participating regulatory authorities. The program was launched by the FDA’s Oncology Center for Excellence (OCE) in 2019 with Health Canada and the Australian Therapeutic Goods Administration.

Since then, five other regulatory counterparts have joined the project, including Brazil's National Health Surveillance Agency (Agência Nacional de Vigilância Sanitária), Israel’s Ministry of Health, Singapore’s Health Sciences Authority, Switzerland’s Swissmedic, and the United Kingdom’s Medicines and Healthcare products Regulatory Agency.
Notably missing from that group has been Japan’s Pharmaceuticals and Medical Devices Agency (PMDA), although it has been involved in oncology monthly cluster teleconference calls with other regulatory agencies since January 2014. However, Japan recently signaled that its involvement in Project Orbis could be changing. In November 2023, an FDA delegation led by OCE’s Director Richard Pazdur visited PMDA as part of a broader trip to Japan, and PMDA expressed interest in resuming their observation of Project Orbis and of potential joint projects with the FDA.
Last year, an FDA OCE delegation also met with the European Medicines Agency (EMA) in Amsterdam to discuss the EMA joining Project Orbis as an observer. OCE says it will focus on cultivating these new areas of collaboration with EMA and PMDA in 2024.
Project Orbis was envisioned as a way of providing cancer patients in other countries with earlier access to products that otherwise might have experienced a significant delay in regulatory submissions within their own countries, regardless of whether the product had received approval in the United States. Pivotal clinical trials in oncology are commonly conducted internationally, not limited to the country of the drug sponsor. For cancer drugs seeking approval in the United States, new drugs are studied against standard of care and not against a placebo. Therefore, future drug development is benefited if the study drug is compared to the latest, uniform standard of care — a benchmark made possible by establishing global standards of treatment.
Seoul Workshop Outcome: Seeking Harmonization on Artificial Intelligence
Global regulatory organizations have been tasked with working on the convergence and harmonization of regulatory strategies on the use of artificial intelligence (AI) in the development of medical products, according to an outcome statement released at the conclusion of the AI Regulatory and International Symposium, also known as AIRIS. The 4-day workshop, which took place on February 26-29 in Seoul, was co-hosted by the FDA and the Republic of Korea’s Ministry of Food and Drug Safety.

Going forward, the outcome statement said, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use and the International Medical Device Harmonization Forum should:
- Promote the development of medical products and contribute to narrowing the gap between countries and regions through continuous sharing of regulatory information on AI-based medical products.
- Recognize ethical issues arising from the development of AI technology and seek common solutions.
- Work to support the convergence and harmonization of regulatory strategies for AI-based technologies throughout a product’s life cycle (development, use, evaluation, etc.) in addition to securing safety and efficacy.
FDA Commissioner Robert Califf set the tone for this approach in virtual opening remarks. “As we move forward in the development of policies in our own regulatory systems, it’s important for each of our agencies to embrace essential principles of convergence and harmonization of regulatory expectations among nations and across areas of medical product innovation,” he said.
“The harmonization process will bring many benefits, including fostering efficiency, consistency, and standardization of best practices, reducing redundancy, and connecting stakeholders who share common interests to advance them. Together, this helps ensure efficient access to markets, and supports the ultimate goal — faster access for patients to innovative, safe, and effective products.”
The symposium featured international speakers from government agencies, industry, and academia, and focused on the use of AI in a wide range of medical products that included biologics, pharmaceuticals, and medical devices. There were seven sessions in all, covering such topics as transparency, explainability, and bias; technology trends (including machine learning for clinical decision support systems and using AI for safety surveillance); and what direction regulators should go in cooperating on the regulatory aspects of AI to ensure that medical products that use this technology remain safe and effective.
The FDA has been working for years to anticipate and prepare for the challenges of AI and to harness its potential. Since 1995, the agency has received over 300 submissions for drugs and biological products with AI components and more than 700 submissions for AI-enabled devices, according to Califf.
The FDA released more information on its AI efforts in a white paper on March 15. The document describes how the agency’s product centers are working together on the topic.
|
|
|
Back to TOP

Expanding Generic Drug Access and Global Harmonization Emphasized at FDA Forum
FDA experts and international regulatory counterparts met recently to discuss the need to expand access to generic drugs globally. In a February webinar, panelists discussed and provided updates on the FDA and EMA’s Parallel Scientific Advice (PSA) Pilot Program for Complex Generic/Hybrid Products, addressed currently available international engagement opportunities, and explored other topics of interest to the generic drug industry.
The webinar was sponsored by the FDA's Center for Drug Evaluation and Research’s Office of Generic Drugs (OGD), which ensures that Americans have access to safe, effective, and high-quality generic drugs. One way that OGD supports generic drug regulatory activities and scientific processes is by facilitating international engagements.

The PSA program gives sponsors an opportunity to meet with European and U.S. regulators and gain consensus on development questions. Initially established for new drugs and vaccines or gene therapies, the PSA program was expanded in 2021 to include complex generics and hybrid products. Ideally, this FDA-EMA collaboration will allow sponsors to avoid unnecessary or duplicative clinical studies while potentially reaching both markets more quickly. The PSA program also aims to foster harmonization of generic drug approval standards across jurisdictions, with the future possibility of creating a global dossier that could be approved by multiple regulators.
Lei Zhang, Ph.D., OGD’s Deputy Director of the Office of Research and Standards said that when applicants engage in concurrent scientific conversations with both agencies on key issues during the development phase of complex generic drug products/hybrid products, thanks to the PSA program, dialogue between the two agencies increases, which encourages regulatory convergence and shortens the time to drug development and approval.
Established by the FDA in 2021 as quarterly videoconferences, the cluster was the first forum dedicated to generic drug development. The collaborative effort has led to improvements in the understanding of regulatory requirements and increased scientific alignment for generic drug development, said Sarah Ibrahim, Ph.D., OGD’s Associate Director of Stakeholder and Global Engagement.
FDA Issues Import Alert for Food Products with PFAS and Other Chemical Contaminants
On March 19, the FDA issued a new import alert for human food products with detectable levels of chemical contaminants that may present a safety concern to human health. The Import Alert 99-48, Detention without Physical Examination of Foods Due to Chemical Contamination, gives the FDA the ability to help prevent entry of human food products into the U.S. if they are found to be contaminated with a broad range of human-made chemicals including benzene, dioxins and polychlorinated biphenyls (PCBs), and per- and polyfluoroalkyl substances (PFAS), among others.
PFAS, a diverse group of thousands of chemicals that resist grease, oil, water, and heat, are used in many different types of products. PFAS in the environment can enter the food supply through plants and animals grown, raised, or processed in contaminated areas. It is also possible for very small amounts of certain PFAS to enter foods through food packaging, processing equipment, and cookware.
The import alert follows an FDA announcement on February 28 that grease-proofing materials containing PFAS are no longer being sold for use in food packaging in the United States. This means the major source of dietary exposure to PFAS from food packaging like fast-food wrappers, microwave popcorn bags, takeout paperboard containers, and pet food bags is being eliminated.

In 2020, following the FDA’s postmarket safety assessment, the agency obtained commitments from manufacturers of food contact materials to cease sales in the United States of grease-proofing substances that contain certain types of PFAS — the February announcement marks the fulfillment of these voluntary commitments. The FDA also has confirmed that other manufacturers have voluntarily stopped sales of other food contact substances that contain different types of PFAS intended for use as grease-proofing agents in the United States.
Grease-proofing substances are applied on paper and paperboard packaging to prevent the leaking of grease and oil, and to provide water-resistant properties. The substances containing PFAS were applied to fast-food wrappers, microwave popcorn bags, takeout paperboard containers, pet food bags, and other similar types of packaging. While the original commitment letters received by the FDA from manufacturers state that paper/paperboard products containing these food contact substances could take 18 months to exhaust the market supply from the last date of sale, most of the companies have exited the market prior to their original phaseout date. The agency is also working toward a validated analytical method that would allow for monitoring the market for these food contact substances in packaging.
The FDA has taken the approach of assessing on a case-by-case basis whether the type and level of a chemical contaminant found in food may pose a health concern, such that the food is considered to be adulterated in that levels may pose a human health risk. To make that determination, the agency considers factors including, but not limited to, whether there is an established action level or tolerance, how much of the food people typically eat, the level of the contaminant detected in that food, and the toxicity of the specific contaminants.
In 2022, the FDA initiated a targeted survey for PFAS in 81 seafood samples collected at retail and determined that the estimated exposure to perfluorooctanoic acid (a type of PFAS) from certain samples of canned clams from China is likely a health concern. The 81 samples in the survey consisted of clams, cod, crab, pollock, salmon, shrimp, tuna, and tilapia, most of which were imported to the United States. The FDA plans an additional targeted survey of molluscan shellfish this year. The new import alert could be used to refuse entry of foods like seafood contaminated with PFAS.
Specific firms and their food products found with levels of chemical contaminants that could pose a risk to human health may be subject to detention without physical examination under this import alert. To remove a firm/product from this import alert, evidence should be provided to the FDA to adequately demonstrate that the firm has resolved the conditions that gave rise to the appearance of the violation, so that the agency will have confidence that future products will be in compliance.
INO Talks Generic Drug Quality
The FDA’s India Office (INO), in collaboration with subject matter experts from the FDA’s Center for Drug Evaluation and Research (CDER), recently organized three technical workshops highlighting the importance of maintaining quality throughout the life cycle of a generic drug product. The workshops featured speakers from CDER as well as regulators from the Indian government and from industry. The first workshop, in December in New Delhi, was co-hosted by India’s Central Drugs Standard Control Organisation and drew over 100 government regulators representing 18 different states in India. In the same month, INO and CDER collaborated with the Drug Information Association to co-host workshops in Ahmedabad and Hyderabad, respectively, with a combined attendance of more than 400 participants and representatives from 90 pharmaceutical companies.

FDA subject matter experts from CDER, DIA organizer, and FDA INO leadership and organizers at the DIA-USFDA-ANDA Workshop, pictured from left to right: Mr. Gregory Smith (Deputy Country Director, INO), Dr. Utpal Munshi (CDER), Mr. Dhruv Shah (INO), Dr. Sarah Ibrahim (CDER), Ms. Kanchan Patel (DIA), Dr. Sarah McMullen (Country Director, INO), Dr. Sharmista Chatterjee (CDER), Ms. Aditi Thakur (CDER), Dr. Jacquin Jones (INO), and Dr. Peter Capella (CDER).
Nine out of 10 prescriptions dispensed in the United States are generic drugs, and 40% of these products are manufactured in India. Pharmaceutical quality is at the forefront of the FDA’s priorities and INO has been a strong proponent for building quality into pharmaceutical manufacturing systems in India, for strengthening India’s regulatory systems, and for working on implementing policies that harmonize with international standards and regulations.

FDA CDER subject matter experts, FDA India office leadership and staff, India's Central Drugs Standard Control Organisation leadership and team, and workshop attendees from central and state drugs control authorities.
ICYMI — OUR LATEST INTERVIEWS:
MHRA on the Value of Partnership, FDA on Global Tobacco Control
OGPS provides occasional thought pieces on international topics in an ongoing series called From a Global Perspective. Lately, these perspectives have been in the form of timely interviews with leaders from across the FDA as well as with our international colleagues. Examples of each were published in February.

Dr. June Raine, CEO of the U.K.’s Medicines and Healthcare products Regulatory Agency (MHRA) sat down and discussed the vital importance of working in partnership with other regulators. After the U.K. left the European Union and was no longer one of a network of European regulators, MHRA adjusted by reaching out internationally, forming alliances with Australia, Canada, Singapore, and Switzerland, for example.
During the interview, Dr. Raine talked about how international recognition will impact MHRA’s role in the future, the special challenge of medicines and vaccines for use in healthy people, memorable FDA/MHRA collaborations, improving diversity in clinical trials, improving safety, and her concerns about disinformation. It was a two-part conversation. The first part of the conversation took place just before an FDA delegation led by FDA Commissioner Robert M. Califf met with Dr. Raine and other MHRA staff in London. Then, one last question was put to Dr. Raine right after Dr. Califf’s visit.
OGPS also featured a conversation with an FDA leader, Brian King, director of the FDA’s Center for Tobacco Products, following his attendance at a major international meeting — the Conference of the Parties (COP) — in Panama. The Conference of the Parties is the governing body of the WHO Framework Convention on Tobacco Control (WHO FCTC), the first international treaty negotiated under the auspices of the World Health Organization. Developed in response to the globalization of the tobacco epidemic, the treaty was adopted by the World Health Assembly in 2003 and entered into force in February 2005 and has since become one of the most rapidly and widely embraced treaties in United Nations history. The COP currently meets every two years, the latest being COP10 in Panama last month.
In his conversation with OGPS, Dr. King not only offered his perspective on the meeting but discussed the FDA’s broader international engagements on tobacco control and prevention.
Update on FDA’s Evaluation of Plastic Syringes Made in China
The FDA provided an update on its ongoing evaluation of quality and performance issues related to plastic syringes made in China on March 19 – including recommendations and action the FDA is taking to address the issues.
“We remain concerned that certain syringes manufactured in China may not provide consistent and adequate quality or performance, and our evaluation is ongoing. This is an evolving situation, and we will continue to keep the public informed as new or additional information becomes available,” said Jeff Shuren, M.D., J.D., director of the FDA’s Center for Devices and Radiological Health.
After receiving multiple reports from consumers, health care providers, and health care facilities, the FDA decided to evaluate the potential for syringe leaks, breakages, and other problems in November. The syringes were made by three entities in China.
On March 18, the FDA issued warning letters describing the violations related to the sale and distribution of unauthorized plastic syringes made in China that have not been cleared or approved by the FDA for sale or distribution in the U.S. to the following three entities:
The warning letters for Medline Industries, LP and Sol-Millennium Medical, Inc. also concern violations related to quality system regulations for syringe products. Until further notice, consumers and medical professionals were advised to immediately transition away from using plastic syringes manufactured by Jiangsu Caina Medical Co. Ltd. and Jiangsu Shenli Medical Production Co Ltd.
Recommendations for all other plastic syringes made in China while the FDA’s evaluation is ongoing are:
- Check the manufacturing location for syringes you use or have in your inventory by reviewing the labeling and/or outer packaging, or contacting your supplier or group purchasing organization.
- Use syringes not manufactured in China, if possible. At this time, glass syringes, pre-filled syringes, or syringes used for oral or topical purposes are not included.
- If you only have syringes manufactured in China, then continue to use them as needed until you are able to use alternative syringes and closely monitor for leaks, breakage, and other problems.
-
Report any issues with syringes to the FDA.
|
|
BONUS PHOTO ALBUM:
FDA Delegation Visits Regulators in Europe
An FDA delegation led by FDA Commissioner Robert M. Califf, M.D., traveled to Europe in February and met with representatives from industry, academia, and civil society, as well as regulators in the United Kingdom, Belgium, and the Netherlands.
On February 19 the delegation met with CEO Dr. June Raine and other officials of the Medicines and Healthcare products Regulatory Agency (MHRA), the U.K.'s regulator of medicines, medical devices and blood components for transfusion. The visit reinforced and deepened the FDA’s and MHRA’s long-standing collaborative relationship.

The two regulatory authorities shared ways to strengthen supply chains and jointly support diversity and inclusion in clinical trials, discussed how to better align on standards and infrastructure to support the secondary use of health data for research purposes, and agreed to work together to combat misinformation and disinformation.
On February 21, the delegation met with Stella Kyriakides, European Commissioner for Health and Food Safety, and others in the Directorate General for Health and Food Safety, or DG Sante. DG Sante has executive responsibility for the regulation of food, feed, veterinary medicines, drugs, biologics, medical devices, and tobacco products in the EU.

During the meeting with Commissioner Kyriakides and her staff, Dr. Califf and FDA leaders relayed the agency’s vision for a single high global standard for medical product quality and talked about strategies for combating misinformation/disinformation. It was also an opportunity to discuss shared priorities such as strengthening supply chains, the U.S.-EU Mutual Recognition Agreement, and heath data governance.

In addition, the FDA delegation met with the European Food Safety Authority (EFSA), the EU agency that provides scientific advice and communicates on risks associated with the food chain. The discussion focused on areas in which the FDA and EFSA can better coordinate on emerging technologies that inform risk assessment practices.

Dr. Califf wrapped up his trip to Europe with a visit to the European Medicines Agency (EMA) in Amsterdam. The EMA, led by Executive Director Emer Cooke, is the EU agency in charge of the evaluation and supervision of pharmaceutical products. The FDA has enjoyed 20 years of close collaboration with the EMA – in fact, the agency has a liaison embedded in the EMA offices in Amsterdam and the EMA has a liaison embedded at the FDA's headquarters in White Oak, Maryland. FDA officials came away from the meeting with Executive Director Cooke and her staff with a robust plan for future collaborations.

|
|
BONUS PHOTO ALBUM:
Meetings with Foreign Delegations in the Washington Area
On March 7, FDA Commissioner Robert M. Califf and other FDA officials met with representatives from Israel’s Ministry of Health at the FDA’s White Oak, Maryland, headquarters to discuss current and future collaborations.

On March 13, FDA Commissioner Robert M. Califf and other FDA officials met with a delegation from India led by Rajiv Wadhawan, Joint Secretary of the Ministry of Health and Family Welfare (fifth from right), and Dr. Rajeev Singh Raghuvanshi, Drug Controller General of India (third from right), who oversees the Central Drugs Standard Control Organisation. The meeting, held at the FDA’s White Oak, Maryland, headquarters, touched on drug quality and international harmonization.

The FDA signed a confidentiality commitment with Kenya's Pharmacy and Poisons Board (PPB) on March 15 in Washington, D.C. The signatories were FDA Associate Commissioner for Global Policy and Strategy Mark Abdoo and Dr. Fred Siyoi, CEO of the PPB. A confidentiality commitment sets up the legal framework for the FDA to share certain kinds of nonpublic information with its counterparts in foreign countries and international organizations. The March 15 documents, one for the FDA, the other for PPB, authorized the sharing of nonpublic information about the medical devices they regulate.

|
|
|
Back to TOP

Recent communications from OGPS to our international stakeholders (list does not include twice-weekly FDA Roundup summaries), February 22, 2024, through March 11, 2024.
|
|
|
Back to TOP

|
March 31
|
OGPS Fifth Anniversary
|
|
April 7
|
World Health Day
|
|
April 22
|
Earth Day
|
|
April 24-25
|
FDA-EU Bilateral Meeting, White Oak, Maryland
|
| |
|
|
|
|
Read thought-provoking pieces covering international topics in
From A Global Perspective.
|
|
|
Please follow @FDA_Global.
|
|
|
Subscribe to Global Update
Don't miss out on future issues of Global Update or other international news from OGPS. To sign up, click "Manage Subscriptions" in the below footer then follow the prompts and select the "International Programs" box.
|
|
|
|
|




