Dr. Heather M. Blankenship, PhD
The first sequenced isolate of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) from a patient in Wuhan, China was shared on 10 January 2020. Since then, over 128,000 SARS-CoV-2 sequences have been generated in the US. SARS-CoV-2 is an RNA virus that belongs to a family of viruses which includes pathogens such as SARS-CoV and MERS-CoV, along with viruses that cause the common cold. All viruses, regardless of the family that they belong to, require a host cell to infect, to undergo replication, and to generate more viruses. It is during this process of replication that mutations arise in the genome. While the mutation rate for RNA viruses tends to be very high, SARS-CoV-2 only generates 2 mutations per month on average, which is half the rate of influenza and a quarter the rate of HIV. However, not every mutation will provide an advantage to the virus and most mutations will go undetected since they result in either no changes or a non-viable virus.
The use of molecular epidemiology to respond to outbreak investigations and provide epidemiological support has been utilized for a range of bacterial and viral pathogens. More recently, whole genome sequencing in the field throughout the Ebola outbreak allowed for the identification of cases to provide real-time interventions and information about the evolution of the virus. Building from the knowledge that we have gained from outbreak interventions of other viral pathogens; scientists have been able to apply the knowledge and methods to the SARS-CoV-2 pandemic. A global community established through the SARS-CoV-2 Sequencing for Public Health Emergency Response, Epidemiology and Surveillance (SPHERES) consortium has coordinated efforts from research, industry, state, and federal partners to respond to SARS-CoV-2 through whole genomes sequencing.
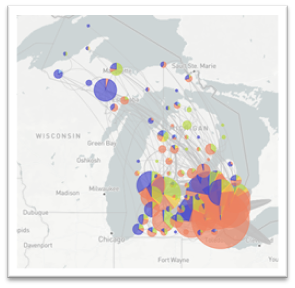
Within Michigan, whole genome sequencing of SARS-CoV-2 has helped to understand the virus and provide support to the pandemic response. By examining the virus at the genomic level along with the date of infections we can generate broad transmission dynamics to watch how the virus might be moving around the state or across state borders (see figure 1). On a smaller scale, we can examine and provide support to epidemiologists as they investigate cluster outbreaks within different facilities by examining slight differences in the genome. While examining the SARS-CoV-2 genomes, we also monitor and track variants that may be of clinical or diagnostic importance. Specifically, variants that may result in more severe clinical outcomes or affect our ability to accurately identify a positive specimen. As we continue to respond to the SARS-CoV-2 pandemic, whole genome sequencing will help provide insight on changes that we see within the virus and how that might affect the response.
Figure 1: Broad transmission dynamics of SARS-CoV-2 in Michigan